
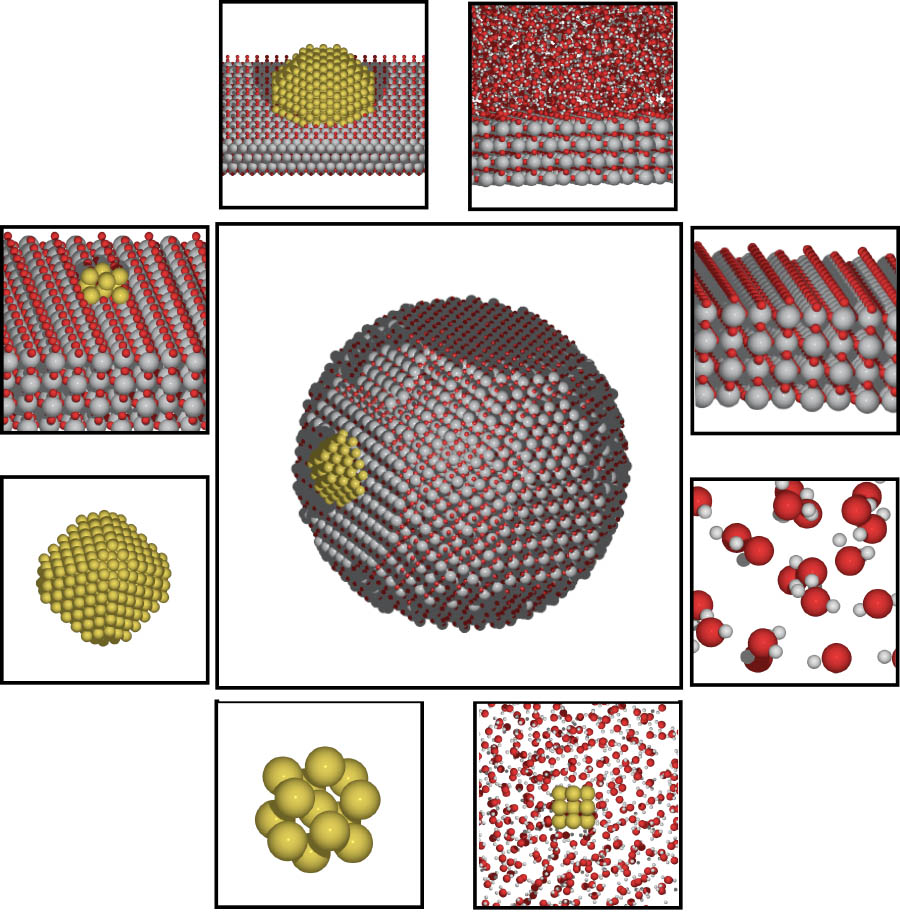
 An illustration depicting chemical catalysis on surfaces and nanostructures. A new $2.8 million study led by School of Civil and Environmental Engineering Associate Professor Phanish Suryanarayana will harness the power of future supercomputers to understand the interactions that take place in these kinds of chemical reactions. (Image Courtesy: Andrew Medford) |
Early in the next decade, the first computers capable of at least one quintillion calculations per second will come online at Argonne National Laboratory.
That’s a one followed by 18 zeroes, or what scientists call “exascale” machines. These will be machines with one billion processing cores. Thing is, we don’t have computer codes that can actually use all that power efficiently — power that has the potential to unlock all kinds of new knowledge.
Phanish Suryanarayana in the School of Civil and Environmental Engineering is leading a team on a new project to make use of all those processors to study the interactions of atoms using quantum mechanics, building on computer code his team has developed in recent years. Funded by the U.S. Department of Energy, the four-year, $2.8 million study — if everything goes well, as Suryanarayana puts it — will mean scientists can study and understand chemical systems that include up to 10 million atoms.
Right now, the best we can do is in the range of about 1,000 atoms.
|
“It will enable us to do applications that have not been possible before,” said Suryanarayana, an associate professor in the School. “I think the number of applications is just limited by our creativity, in some sense.”
Suryanarayana has partnered with Andrew Medford, assistant professor in Georgia Tech’s School of Chemical and Biomolecular Engineering; Polo Chau and Edmond Chow, associate professors in the School of Computational Science and Engineering; and John Pask, a physicist at Lawrence Livermore National Laboratory. Their project is one of only 10 awards for the development of new software to design new chemicals and chemical processes. The Energy Department said software that comes out of these studies should lead to the development of new solvents, photo-sensitive materials, and catalysts, for example.
That’s where the Georgia Tech project comes in, which will focus on what Suryanarayana called dynamic catalysis.
“In other words, we want to propagate in time to see which atom goes where [in a reaction], which interacts with what, and how the phenomenon actually occurs,” he said.
 Chau |
 Chow |
 Medford |
 Suryanarayana |
“[The code we’re working on] allows us to now model systems, largish systems, which are representative of practice, and watch them evolve in time to see what's happening and identify those sites where the catalysis is happening — to actually design new catalysts, better, more efficient ones.”
Those catalysts could have applications in everything from making chemical reactions in fuel cells more efficient to improving combustion or the catalytic converters in automobiles.
Suryanarayana said many catalytic phenomena are poorly understood because of the complex bonding that happens in a chemical reaction. They are also dynamic — they occur over time — which means computer models have to process many millions of data points many thousands of times.
“We need to see what's happening in this chemical reaction that evolves in time: the atoms are moving around based on forces and their reactions, and the configuration keeps changing,” he said. “You actually need quite a few steps [in the computer simulation]. For example, you might need 10,000 steps or 100,000 steps.”
Running that many steps even for 1,000 atoms in a simulation adds up quickly, both in terms of time and costs. The goal with the code the research team will develop is to cut that time drastically while also producing highly accurate results.
To do it, they’ll have tackle three confounding problems.
First, creating an algorithm that will actually function efficiently on a billion different processors simultaneously.
It turns out, this kind of quantum mechanical simulation can’t even use the full power of current petascale supercomputers operating around a million processing cores, Suryanarayana said. The most advanced algorithms today can use perhaps 10,000 cores.
Second, when you scale from modeling, say, 100 atoms to modeling 1,000 atoms, the time increases cubically. So not 10 times longer to run the simulation, but 1,000 times longer.
And third, the sheer number of data points needed to capture and understand atomic interactions means that the models have to account for a lot of unnecessary information.
Suryanarayana said his team recently discovered a new approach that eliminates a lot of the unneeded data, making the computations significantly more efficient.
“You don't know what you're searching for [in the simulations], so you need to incorporate the ability to capture more information than it actually requires. Our method does the additional step of getting rid of the unwanted stuff by utilizing the knowledge of the local chemistry of the system,” he said. “Initially you might have 10,000 basis functions per atom, roughly; this reduces it to like 10 or 20.”
The team also will use machine learning to further accelerate the computations. And they’ll use quantum mechanics work Suryanarayana’s group has done creating algorithms that scale up without the huge increase in computational time.
The potential, he said, is enormous, considering the amount of new science created last year using one of the top codes currently available.
“That code was used around 7,000 times; it got 7,000 citations last year [in scientific papers],” Suryanarayana said. “Seven thousand times the code was used and actually got new science, and that code only goes up to about a thousand atoms. We're looking at building a code for 10 million atoms.”